|
A Mixture of Feature
Experts Approach for Protein-Protein Interaction Prediction Yanjun Qi1, Judith Klein-Seetharaman1,2, Ziv
Bar-Joseph1 1School of Computer Science, 2Department of Structural Biology, |
|
Abstract Background: High-throughput methods can directly detect the set of interacting proteins in model species but the results are often incomplete and exhibit high false positive and false negative rates. A number of researchers have recently presented methods for integrating direct and indirect data for predicting interactions. These methods utilize a common classifier for all pairs. However, due to missing data and high redundancy among the features used, different protein pairs may benefit from different features based on the set of attributes available. In addition, in many cases it is hard to directly determine which of the data sources contributed to a prediction. This information is important for biologists using these predications in the design of new experiments. Results: To address these challenges we propose a Mixture-of-Feature-Experts method for protein-protein interaction prediction. We split the features into roughly homogeneous sets of feature experts. The individual experts use logistic regression and their scores are combined using another logistic regression. When combining the scores the weighting of each expert depends on the set of input attributes available for that pair. Thus, different experts will have different influence on the prediction depending on the available features. Conclusions: We applied our method to predict the set of interacting proteins in yeast and human cells. Our method improved upon the best previous methods for this task. In addition, the weighting of the experts provides means to evaluate the prediction based on the high scoring features. PDF version (PDF) Online version
(Online) |
||||||||||||||
|
Features (Yeast PPI) ·
For
the various data sources, each of them has its own representative form. For example,
protein sequence is in the form of a character string, which means the order
of amino acids as they occur in a polypeptide chain. Gene expression data is
usually a vector of expression values across multiple time points for a
specific gene. Synthetic lethal data describes that a pair of genes having
mutations together would affect the cells inviable
or viable. So how could we combine these different forms of data together? We
present the converting process briefly in Figure 1. For each data set that
represents a certain gene / protein's property, we figured out one natural
way to calculate the similarity between two genes / proteins with respect to
the specific evidence. For instance, for two proteins' sequence information,
we use BlastP sequence alignment E-value as one
feature for this protein-protein pair from the protein sequence evidence. For
other data sources, similar procedures were pursued to make the features for
a protein pair. Concatenating these features together then gave us the feature
vector describing a protein-protein pair. ·
The
yeast features used in the paper were described in detail in the following
paper o
Y. Qi, Z. Bar-Joseph, J. Klein-Seetharaman, "Evaluation
of different biological data and computational classification methods for use
in protein interaction prediction", PROTEINS: Structure, Function, and Bioinformatics. Jan
2006 o
All
related data sources and how they were converted into features representing
pair of proteins have been described in details in: http://www.cs.cmu.edu/~qyj/papers_sulp/proteins05_pages/features.html o
These
data sets could also be downloaded from: http://www.cs.cmu.edu/~qyj/papers_sulp/proteins05_pages/feature-download.html
|
||||||||||||||
|
Features (Human PPI) ·
The
human PPI features used in the paper were described in detail in the paper ·
We
share one run of the features and their related pairs @ the following ULR: o
http://www.cs.cmu.edu/afs/cs.cmu.edu/project/structure-9/PPI/mfe07-HumanGeneral/
|
||||||||||||||
|
Discussion of Number of Feature Experts ·
As
discussed in the 'Methods' section, the number of feature experts that our heterogeneous
data sets are split into could be different to what we used in the paper. The
splitting depends on the need of the application and the preference of the
biologists who would analyze and/or validate the predictions. ·
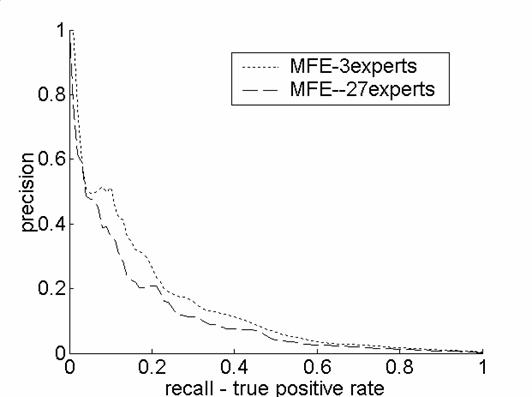
The
methodology we propose, of feature experts, is very general. At the limit we
can assign each feature to an individual expert. To test this we carried out
new experiment for the human prediction task treating every feature as its
own expert. As the following supporting Figure 1 indicate, this does not
improve the performance of the algorithm we present, perhaps because it leads
to overfitting of the parameters. ·
Supporting Figure 1:
·
Supporting Table 1:
|