Back to "Biological Language Modeling Seminar Topics"
General reference: Ref_JianEtal Chapter 16
Protein folding
The native state is characterized by sterically allowed conformations that energetically favorable, see chapter "ab initio protein structure prediction".
Sampling of all the possible conformations and deciding which conformation conforms to the above steric and energetic conditions would take forever (Levinthal paradox).
The folding pathway occurs via unstable intermediates (unstable because even the native state is so marginally more stable than the unfolded state). One such intermediate is the molten globule, a conformational ensemble with some native secondary structure elements, but lacking tertiary interactions that lock the protein in the native state. This means that local interactions are useful in providing a low-energy pathway for structure assembly. Non-native interactions generally slow down the folding process, but can also prevent irreversible misfolding due to the formation of other non-native contacts that may prevent productive folding.
Ideally, simulations of all-atom models would provide an understanding of the driving forces behind the folding process, however the CPU time needed is is many orders of magnitude more than the physical time simulated (Daggett 2000). The longest simulation of any protein folding process has been Duan and Kollman's 1998 study of the 36aa villin headpiece (9295 atoms), tracking a single dynamic trajectory that corresponds to 1 microsecond of physical time.
7aa beta-peptide in methanol has been successfully folded by MD simulation to its experimentally determined stable conformation (Daura et al. 1998).
Even the fastest folding proteins take tens of microseconds to fold - at least 10 times longer than the longest simulation to date.
Problems:
1. empirical force fields are not universal and need to be optimized for particular applications.
2. only individual MD trajectories possible - small number of detailed information does not provide overall picture.
Approximations:
Coarse-grained models.
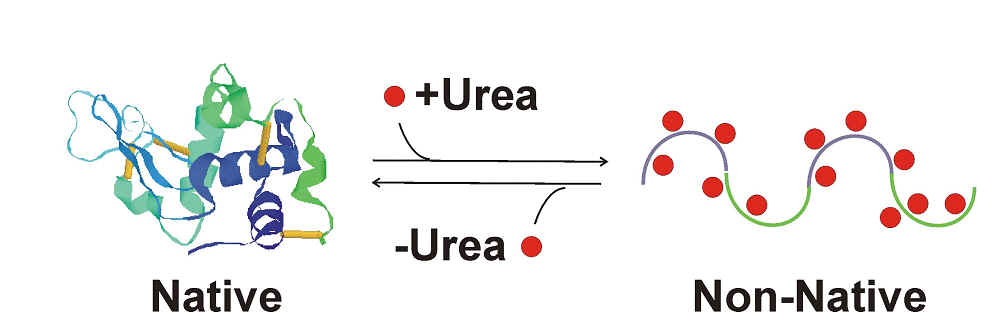
Figure: Experimental approach to the study of protein folding: denaturation and renaturation of folded proteins
Misfolding is the cause of many diseases. Examples: Alzheimer, BSE, familial amyloidosis, Creuzfelt-Jacob disease, Retinitis pigmentosa, Cystic fibrosis
Misfolding occurs because of 1. Changes in the protein sequence 2. Change in conformation of the same sequence
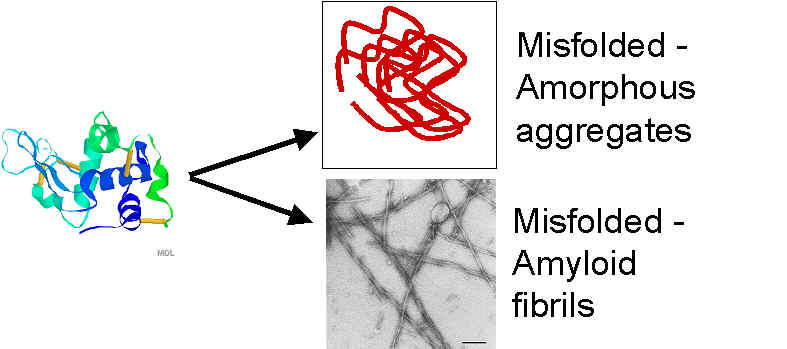
Figure: Misfolding
Lattice models of protein folding
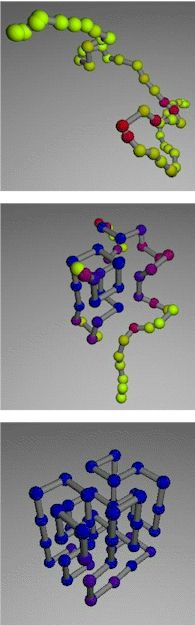
Figure: unfolded (top), intermediate (middle) and stable conformations of a polymer (bottom). Taken from http://www.lbl.gov/Science-Articles/Archive/model-protein-folding.html PNAS 1999 paper
Review Schwartz and Sorin Istrail papers with Jonathan King - how lattice models can model misfolding as well. Lattice models have been used to model folding, unfolded, intermediate structures. Folding pathways and folding kinetics. Low resolution folded models.
Two Basic Principles of Protein folding
see Mirny and Shakhnovich, 1999
1. Foldability Principle
= the native conformation is a pronounced energy minimum
- sequences that satisfy this requirement
- fold fast
- have cooperative folding transition
- native structures are stable against mutations
- native structures are stable against variation in solved conditions and temperature
2. Nucleation in folding kinetics
- obligatory contacts (specific nucleus) should be formed in order for a protein to reach transition state
- folding nucleus is a spatially contiguous cluster in structure, but not necessarily in sequence: non-local contacts are present
- after forming nucleus, folding is fast and downhill
- location of a folding nucleus depends on structure more than on sequence