Using expression QTL for human disease gene mapping
Traditionally genetic studies focus on the correlation between genetic variations and organism-level phenotypes. The emerging field of systems genetics studies how DNA variations affect molecular phenotypes, such as gene expression, metabolites, and DNA methylation, which provide a bridge from genotypic changes to phenotypes. A large and growing number of system genetic studies have been performed, often focusing on gene expression traits (called eQTL studies). I built a statistical framework, named Sherlock, to jointly analyze eQTL and data from genome-wide association studies (GWAS). This method allows us to effectively combine many weak signals in GWAS to identify disease susceptibility genes. Because many such signals are linked to expression of a gene in trans, Sherlock is able to detect completely new genes from GWAS, and we made promising discoveries in a range of different diseases.
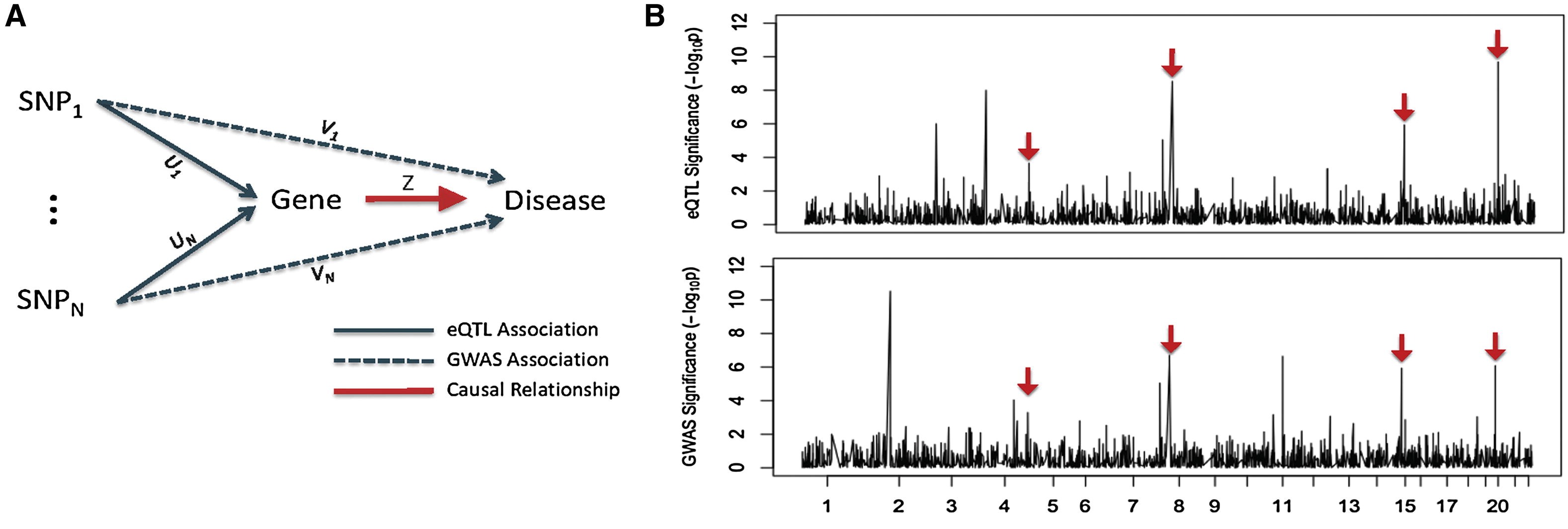
- Sherlock: Detecting Gene-Disease Associations by Matching Patterns of Expression QTL and GWAS.
He X, Fuller CK, Song Y, Meng Q, Zhang B, Yang X, Li H. Am J Hum Genet, 2013 May 2;92(5):667-80 (Featured by Am. J Hum. Genet. and Genetics).
Identifying risk genes by using rare genetic variations
Each person inherits mutations from parents, some of which may predispose the person to certain diseases. Meanwhile, new mutations may occur spontaneously during the reproductive process, and if disrupting key genes, these de novo mutations may increase risks of diseases, especially neurodevelopmental ones. We developed a likelihood model that effectively combines data from multiple sources of the same genes: de novo mutations, inherited variants identified from families, and standing variants in the population (identified with case-control studies). We use a Hierarchical Bayes strategy to borrow information across genes to improve statistical inference. This highly integrative approach greatly increases the power of gene discovery, and predicts promising genes for autism.
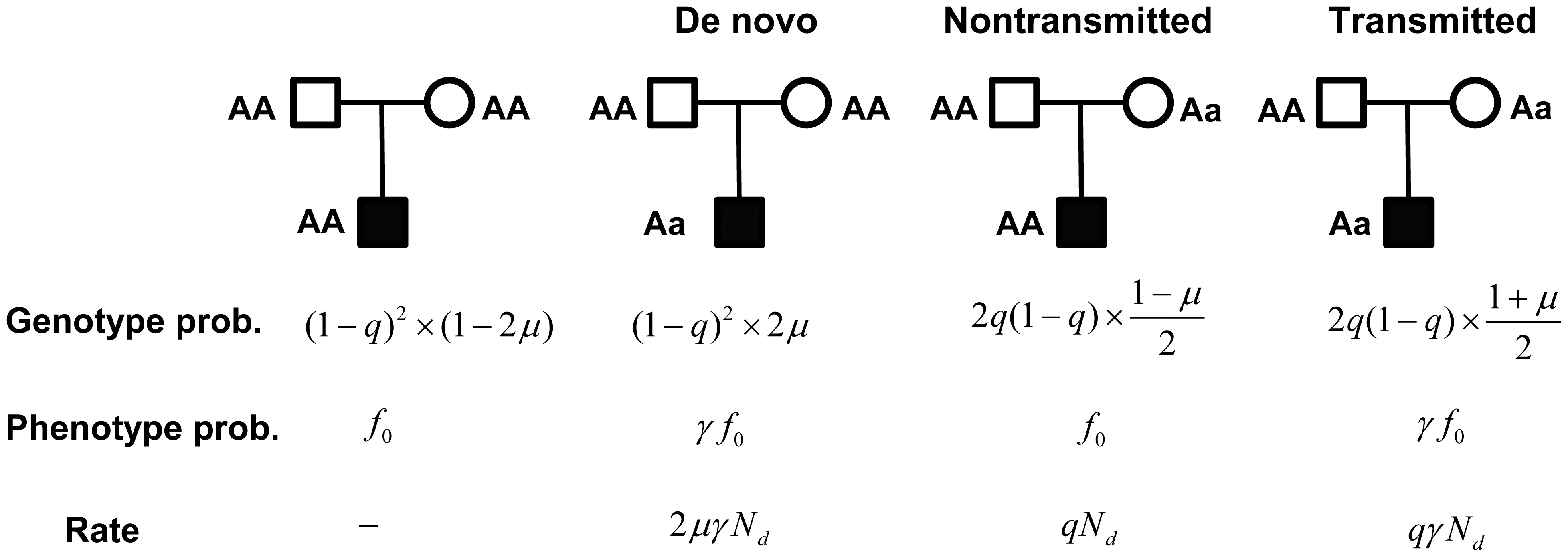
- Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes.
He X, Sanders SJ, Liu L, De Rubeis S, Lim ET, Sutcliffe JS, Schellenberg GD, Gibbs RA, Daly MJ, Buxbaum JD, State MW, Devlin B, Roeder K. PLoS Genetics, 2013 Aug;9(8):e1003671 (SFARI News and Faculty of 1000).
Quantitative modeling of regulatory sequences
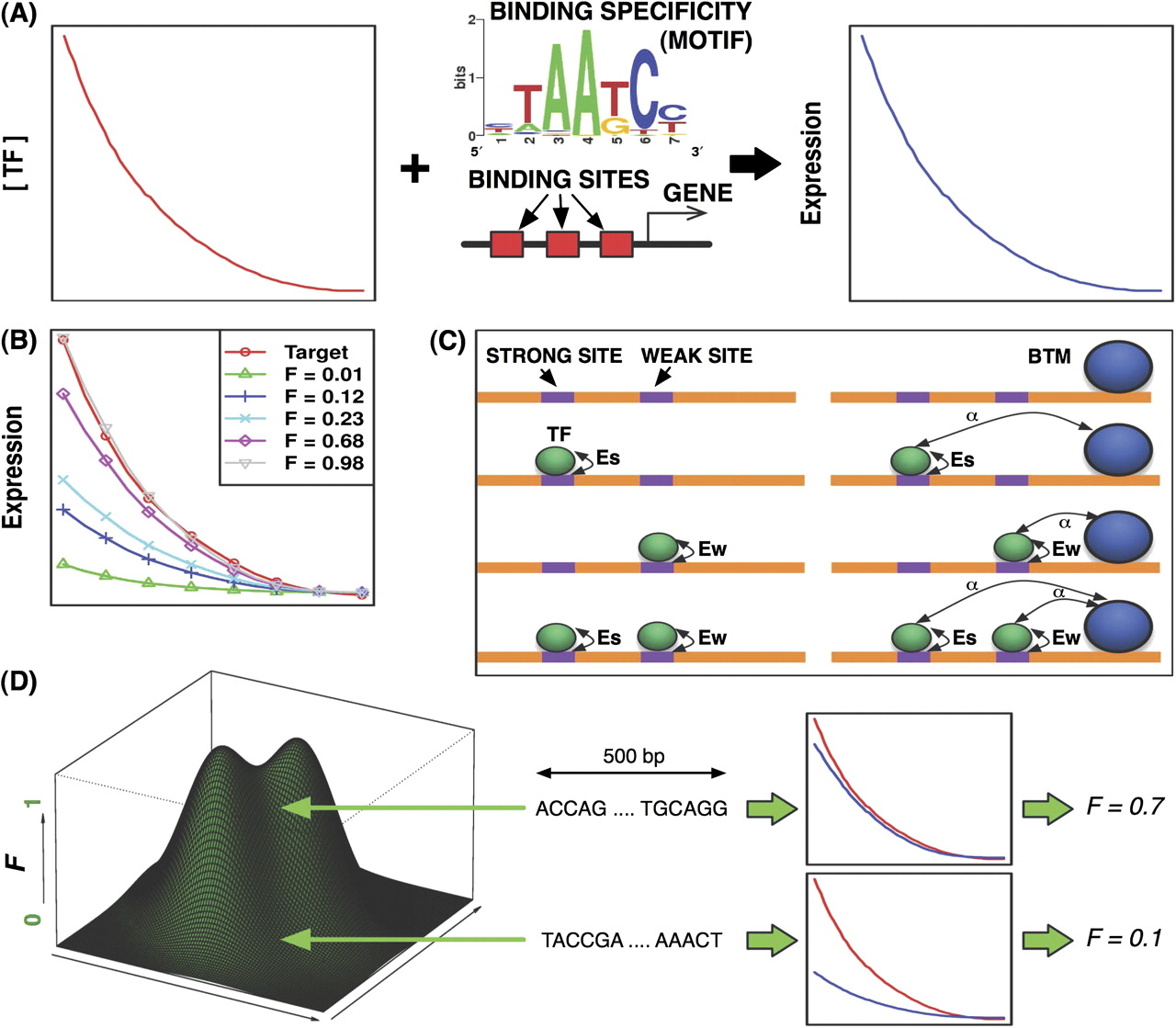
 Regulatory DNA sequences drive gene expression patterns by integrating information about the environment in the form of the activities of transcription factors. The rules by which regulatory sequences read this type of information, however, are unclear. I developed quantitative models based on physicochemical principles that directly map regulatory sequences to the expression profiles they generate. These models incorporate mechanistic features that attempt to capture how activating and repressing factors work together. By evaluating the importance of these features in the fruit fly segmentation system, we were able to gain insights on the quantitative regulatory rules, including the way repressors prevent transcriptional activation, and the role of cooperative interactions. A simpler model was also applied to ChIP-seq data of transcription factors important for embryonic stem cells, and was shown to be significantly more predictive of DNA binding affinities than other existing methods.
Regulatory DNA sequences drive gene expression patterns by integrating information about the environment in the form of the activities of transcription factors. The rules by which regulatory sequences read this type of information, however, are unclear. I developed quantitative models based on physicochemical principles that directly map regulatory sequences to the expression profiles they generate. These models incorporate mechanistic features that attempt to capture how activating and repressing factors work together. By evaluating the importance of these features in the fruit fly segmentation system, we were able to gain insights on the quantitative regulatory rules, including the way repressors prevent transcriptional activation, and the role of cooperative interactions. A simpler model was also applied to ChIP-seq data of transcription factors important for embryonic stem cells, and was shown to be significantly more predictive of DNA binding affinities than other existing methods.
- Thermodynamics-based models of transcriptional regulation by enhancers: the roles of synergistic activation, cooperative binding and short-range repression
He X, Samee MA, Blatti C, Sinha S. PLoS Comput Biol, 2010 Sep 16;6(9). pii: e1000935 - A biophysical model for analysis of transcription factor interaction and binding site arrangement from genome-wide binding data
He X, Chen CC, Hong F, Fang F, Sinha S, Ng HH, Zhong S. PLoS One 2009 Dec 1;4(12):e8155. (RECOMB Regulatory Genomics)
Prediction of regulatory sequences through comparative genomics and integrated method
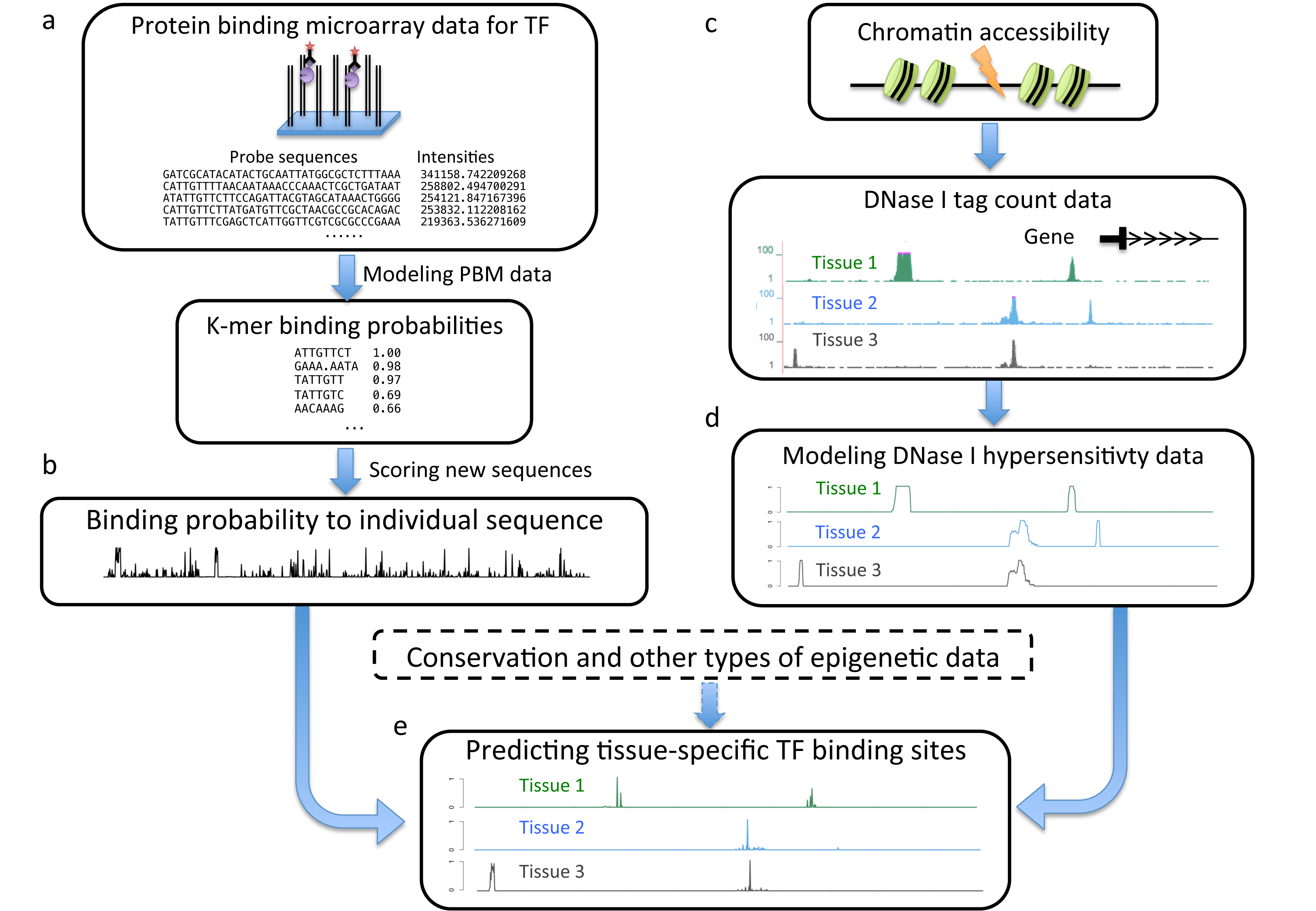 Computational prediction of regulatory sequences may rely on sequence content: whether a sequence contains binding sites that match the specificity of transcritpion factors (TFs). Cross-species genome comparison can further improve prediction because true functional sites tend to be conserved during evolution. We developed computational methods to implement this idea. These methods are built on stochastic models that describe both the sequence content and the evolution of regulatory sequences. In particular, these models capture binding site gain and loss events during evolution. This feature allows our methods to predict partially conserved binding sites, often important in multiple species comparison or comparison of relatively divergent pairs. In a different approach, we built a model to integrate data from in vitro TF binding (protein binding microarray), chromatin accessibility (DNase-seq) and evolutionary conservation. The result is a comprehensive map of binding sites of nearly 200 TFs across 50 different tissues.
Computational prediction of regulatory sequences may rely on sequence content: whether a sequence contains binding sites that match the specificity of transcritpion factors (TFs). Cross-species genome comparison can further improve prediction because true functional sites tend to be conserved during evolution. We developed computational methods to implement this idea. These methods are built on stochastic models that describe both the sequence content and the evolution of regulatory sequences. In particular, these models capture binding site gain and loss events during evolution. This feature allows our methods to predict partially conserved binding sites, often important in multiple species comparison or comparison of relatively divergent pairs. In a different approach, we built a model to integrate data from in vitro TF binding (protein binding microarray), chromatin accessibility (DNase-seq) and evolutionary conservation. The result is a comprehensive map of binding sites of nearly 200 TFs across 50 different tissues.
- Alignment and prediction of regulatory sequences based on a probabilistic model of evolution
He X, Ling X, Sinha S. PLoS Comput Biol, 2009 Mar;5(3):e1000299 - MORPH: probabilistic alignment combined with hidden Markov models of cis-regulatory modules]
Sinha S, He X. PLoS Comput Biol, 2007 Nov;3(11):e216 - Predicting tissue specific transcription factor binding sites.
Zhong S*, He X*, Bar-Joseph Z. BMC Genomics, 2013 Nov 15;14(1):796
Evolution of cis-regulatory sequences
 Understanding the conservation and change of regulatory sequences is critical to our knowledge of the unity as well as diversity of animal development and phenotypes. We tested key evolutionary hypothesis of cis-regulatory evolution using sequence data of more than 50 developmental enhancers across 12 Drosophila species. We made several interesting findings: for example, there are substantial epistatic interactions among different positions of a transcription factor binding site; loss of functional binding sites roughly follows a molecular clock; and the evolutionary fate of a binding site often depends on its sequence context. In another study, I used both theoretical and simulation studies to demonstrate how redundancy (homotypic clustering of transcription factor binding sites) is built into regulatory sequences by evolution, even though redundancy is never directly selected.
Understanding the conservation and change of regulatory sequences is critical to our knowledge of the unity as well as diversity of animal development and phenotypes. We tested key evolutionary hypothesis of cis-regulatory evolution using sequence data of more than 50 developmental enhancers across 12 Drosophila species. We made several interesting findings: for example, there are substantial epistatic interactions among different positions of a transcription factor binding site; loss of functional binding sites roughly follows a molecular clock; and the evolutionary fate of a binding site often depends on its sequence context. In another study, I used both theoretical and simulation studies to demonstrate how redundancy (homotypic clustering of transcription factor binding sites) is built into regulatory sequences by evolution, even though redundancy is never directly selected.
- Evolution of regulatory sequences in 12 Drosophila species
Kim J*, He X*, Sinha S. PLoS Genet, 2009 Jan;5(1):e1000330 - Evolutionary Origins of Transcription Factor Binding Site Clusters.
He X, Duque TS, Sinha S. Mol Biol Evol, 2012, 29(3):1059-70
Genome rearrangement and prediction of functional gene groups
During evolution, the order and relative proximity of genes in genomes are generally not well conserved because of the rapid genome rearrangement events. On the other hand, functionally related genes may be constrained to remain close to each other due to natural selection. Thus, identifying these so-called conserved gene clusters is one way of finding functional gene groups, and can be used to reveal the forces underlying the evolution of genome organization. However, substantial genome rearrangements pose unique computational challenges. I developed a combinatorial algorithm to detect these gene clusters in pairwise genome comparison , allowing genes in the clusters to appear in arbitrary orders. Later, I helped my colleague, Xu Ling, to improve the efficiency of the algorithm, and extend the analysis to a large number of genomes. By combining the algorithmic approach and our newly developed statistical method, we analyzed more than one hundred bacterial genomes and predicted many novel functional gene groups.
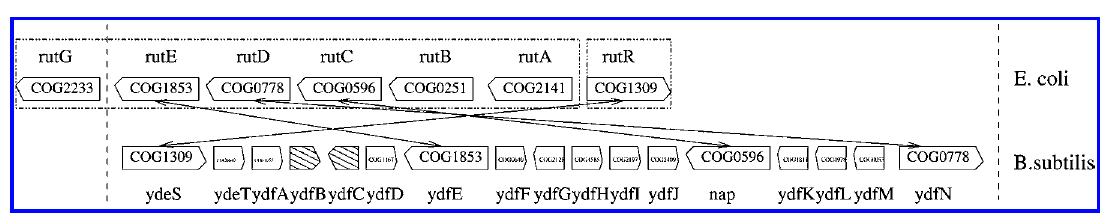
- Detecting gene clusters under evolutionary constraint in a large number of genomes
Ling X, He X, Xin D. Bioinformatics, 2009 Mar 1;25(5):571-7 - Efficiently identifying max-gap clusters in pairwise genome comparison
Ling X, He X, Xin D, Han J. J Comput Biol, 2008 Jul-Aug;15(6):593-609. - Identifying conserved gene clusters in the presence of homology families
He X, Goldwasser M. J Comput Biol, RECOMB Special Issue, 2005 Jul-Aug;12(6):638-56
Biomedical literature mining
Text mining is aimed at extracting information automatically from the vast biological literature. In my RA work with BeeSpace, I was involved in several projects that developed novel text mining methods and practical systems. In one project, we developed the BeeSpace question/answering (BSQA) system that performs integrated text mining for insect biology. BSQA recognizes a number of entities and relations, from gene interactions to insect behavior, in Medline documents. For any text query, BSQA is able to automatically identify important concepts associated with this query, arranged in different categories. By utilizing the extracted relations, BSQA is also able to answer many biologically motivated questions, from simple ones such as, where is a gene expressed, to more complex ones involving multiple types of relations. In another project, I proposed a new statistical method that mines biological literature to find important concepts characterizing sets of genes.
- BSQA: integrated text mining using entity relation semantics extracted from biological literature of insects
He X, Li Y, Khetani R, Sanders B, Lu Y, Ling X, Zhai C, Schatz B. Nucleic Acids Res, 2010 Jul 1;38 W175-81. - Identifying overrepresented concepts in gene lists from literature: a statistical approach based on Poisson mixture model
He X, Sarma MS, Ling X, Chee B, Zhai C, Schatz B. BMC Bioinformatics, 2010 May 20;11:272.