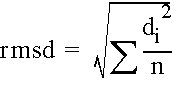
Back to "Biological Language Modeling Seminar Topics"
Back to "Protein structure prediction"
Measures of structural similarity
1. RMSD
- Root mean square deviation of superimposed atoms of two structures
- most widely used
where di is the distance between the ith pair of points after optimal fitting and n is the number of points.
There are three important cases of treating structural alignment with increasing complexity in their determination method:
1. Structure alignment when the correspondence of atoms is known:
pi <--> qi, i=1,...,N
2. Structure alignment when the linear sequence of the residues restrict the possibilities of alignment
pi(k) <--> qj(k), k=1,...K < N,M, where k1>k2 and i(k1) > i(k2) and j(k1) >j(k2)
3. Structure alignment with entirely unknown correspondence.
pi(k) <--> qj(k)
This case is important when identifying a pharmacophore (=a common constellation of a relatively small subset of the atoms of two molecules (e.g. proteins), which are responsible for the biological activity). Task: to find the maximal subset of atoms from two molecules with similar structure.
2. Levitt and Gerstein structural alignment score:
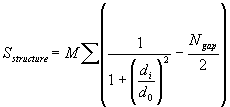
M and d0 are constants set at 10 and 5 A, Ngap is the number of gaps in the alignment, di is the distance between each aligned pair of Calpha atoms, and sum over all aligned pairs i
Advantage over RMSD: di is in the denominator, i.e. the smallest distances are the most significant in determining the score, thereby no need for trimming
3. Distance and contact maps
reviewed in Mizuguchi and Go (1995) Seeking significance in three-dimensional protein structure comparisons. Curr. Opin. Struct. Biol. 5, 377-382. Get ref.
4. Protein structural distance (PSD)
- reference Yang and Honig (2000) JMB 301, 665-678.
- designed as a measure of structural similarity that correlates with sequence similarity, i.e. for pairs of sequences with low sequence identity twilight zone"
- important for homology modeling and fold recognition, where correct sequence to structure alignments are bottleneck
- good correlation between RMSD and sequence similarity for proteins with significant levels of sequence identity (Chothia and Lesk, 1986)
- only useful for RMSD values less than 5 A
- what to do when RMSD is greater than 5A? This is why PSD was developed.
- PSD is applicable for both, high and low similarity
- PSD is implemented in PrISM (protein informatics system for modeling)
- Principle:
structural alignment procedure that uses double dynamic programming to align secondary structure elements and an iterative rigid body superposition that minimizes RMSD of Calpha
the RMSD is good for closely related structures
the SSE is good for distantly related structures
- Result:
continuous conformation space, rather than discrete classification
fully automated procedure to quantify structural similarity (similar to Z-score in approach, dissimilar to SCOP, because it is manual and uses functional information too)
significant overlap with SCOP, but some discrepancies found
- Use of PSD score to establish link between protein sequence and protein structure similarity when sequence identity is <25% (Yang and Honig (2000)):
- sequence identity = ration of the number of identical residues in a pair of aligned sequences to the length of the shorter of the two sequences
- monotonic increase in RMSD down to 25% sequence identify, where the average RMSD is 2A (Chothia and Lesk, 1986, Russell adn VBarton, 1994, WIlson et al., 2000, Wood and Pearson, 1999)
- PSD score of 2 and higher means that the sequences are likely not related
- PSD score of 2 and lower means that there will also be sequence-relationships as defined, not by sequence identity, but rather more sensitive NSWS (= normalized with respect to substitution matrix and peptide length Smith Waterman score. ref. Karlin and Altschul, 1990 and 1993)
- PSD similarities and sequence patterns (albeit not necessarily discernible) are non-random at levels that are below random noise level of sequence alignment scores
4. Z-score
FSSP/Dali measure of structural similarity
Structural alignment Methods:
* DALI, Distance matrix alignment (Holm and Sander, 1995)
* SSAP (Taylor and Orengo, 1989)
* VAST (Gibrat et al., 1996)
Classification of protein structures:
* SCOP, Structural classification of proteins
* CATH, Class, Architecture, Topology, Homologous Superfamily
* FSSP/DDD (fold classification based on structure-structure alignment of proteins/Dali domain dictionary), quantifies structural similarity by Z-score
* CE, Combinatorial extension method
How many distinct folds are there?
- Chothia (1992) Proteins. One thousand families for the molecular biologist [news]. Nature 357, 543-544.
- Orengo et al. (1994) Protein superfamilies and domain superfolds. Nature 372, 631-634. get ref.
- at the moment 700 folds (see SCOP)
- predicted: 1500 soluble proteins (Jones & Hadley in Ref_HigginsTaylor)
- estimates have ranged from 1,000 to 10,000 [see Taylor, W.R. (2002) A 'periodic table' for protein structures. Nature 416, 657-660.]
- estimated 1000-7000 different protein families without sequence similarity [L. Holm and C. Sander (1996) Mapping the protein universe. Science 273, 595-603]
How can the same 3d structure be encoded by the seemingly unrelated protein sequences? OR: What are the numbers of critical residues for protein folding?
- two possible answers:
1. structural information is contained in only a small subset of critical residues and that conserved features are so sparsely distributed in the sequence that they cannot be detected with pairwise sequence alignment. If correct, one wants to identify these critical residues using structure-based sequence profile.
2. 3d information is encoded in different locations throughout the sequence and that critical residues need not be located in the same position in different proteins of the same fold
- Goals: understand protein folding by correlating folding principles to sequence
- Studies addressing these alternatives:
1. Ptitsyn (1998) seven subfamilies of c-type cytochromes:
4 positions besides Heme-coordinating residues are conserved
these 4 position are part of a network of conserved contacts between N-terminal and C-terminal helices
the docking of these 2 helices is early folding step
conclusion: folding of the cytochromes involves the formation of the same conserved folding nucleus
the 4 positions lie on helices, so secondary structure propensities would be important in addition to the conserved 4 positions
critique: sequence conservation could be evolutionary rather than structure-based
2. Bashford et al. (1987) 226 globins:
large number of conserved residues
some of the most conserved residues are exposed to solvent and do not make contacts between helices
all helices have conserved residues, not just the helices known to be involved in the folding nucleus
conclusion: residues other than those involved in folding nucleus can be important determinants of 3D structure
critique: sequence conservation could be evolutionary rather than structure-based
3. Bork et al. (1994) Immunoglobulin-like domains with <25% pair wise similarity
based on structure alignment by DALI
result: absence of common sequence pattern or localized set of interactions that could be crucial determinants to the Ig-like fold
# and location of disulfide bonds variable
conclusion: maybe the Ig-like fold has different folding pathways
4. Halaby et al. (1999)
no common sequence signature for an Ig-like fold
despite importance of the hydrophobic core, there is no strict conservation of the location of individual hydrophobic contacts
5. Russell and Barton (1994) 607 pairs of proteins with similar folds
multiple structural alignment
at the time, there were 2000 different proteins with known structure, in 150 unique fold families
comparison of
side-chain pair contacts: as few as 12%
accessibility (buried, half-buried, exposed): as few as 30% in same category
secondary structure: as few as 41% secondary structure identity (= random)
RMSD 3.2A
only 30% in common core
conclusion: no small number of crucial interactions that are characteristic of a particular fold
6. Mirny and Shakhnovich (1999) Conservatism of conservatism in 5 structural superfolds: immunoglobulin, oligonucleotide-binding, Rossman, alpha/beta plait, TIM barrel folds
identify positions that are conserved within each family and coincide when non-homologous proteins are structurally superimposed
conservation of sequence and structure in 5 structural superfolds ("conservatism of conservatism"): 2200 domains in known structures without evident sequence homology, 564 belong to these 5 dominant folds
well-defined locations in a structure that are crucial for a particular fold
hypothesis: different (even non-homologous) sequences that fold into the same structure may have similar folding nuclei
reasons for conservation
function
stability
historical reasons (insufficient time to diverge)
evolutionary pressure towards fast folding
how to distinguish between different reasons for amino acid conservation?
approach: analyze protein sequences with common fold but not function (no homologous sequences): conserved positions related to structural stabilty and folidng kinetics rather than function
method: structural alignment based on FSSP database (from DALI) and sequence alignment from HSSP database
degree of evolutionary conservation within family of homologous sequences measured by sequence entropy:
Intrafamily conservatism:
![]()
where pi(l) is frequency of each of the six classes i of residues at position l in the multiple sequence alignment
Six classes:
aliphatic AVLIMC
aromatic FWYH
polar STNQ
positive KR
negative DE
special GP
low value of intrafamily conservatism s(l) indicates evolutionary pressure
Conservatism-of-conservatism CoC:
![]()
where l is position in the structural alignment and sm(l) is intrafamily conservatism in family m
- results:
in all folds, residues that show high CoC form a dense cluster in the native structure
the interactions at the key topological positions can be different (e.g. a disulfide bond in one sequence family and a hydrophobic contact in another in the immunoglobulin fold)
correlated mutations exist, but usually involve more than 2 residues
- for 3 of the 5 superfolds (immunoglobiulin, Rossman and alpha/beta plaits), the high CoC is related to folding nucleus
- in some cases, high CoC residues are part of the active site, i.e. in TIM barrels and Rossman fold which are mostly enzymes: speculation that the active sites needs to be dynamically restricted to allow catalysis to occur: advantageous to place active sites near folding nuclei to ensure sufficient stability of the active site against thermal fluctuations
7. Yang and Honig (2000) PSD method applied to 9 superfolds on sequences with <40% sequence identity
- method: generate structure-based sequence profiles for nine superfolds automatically (without using sequence information), then identify sequence patterns in each subset
- use PSD threshold to avoid a dilution effect on information content
- result: most conserved residues are located where tertiary interactions occur and strong structure conservation, but overall patterns are weak and structures can fold without a set of highly conserved clusters or profiles at all (Figure 12, Yang and Honig, 2000). Types of conservation differs in different superfolds
- conclusion: structure-determining factors that do not require specific sequence are important to encode protein fold
- secondary structure propensities
- hydrophobic interactions
- conclusion: high sequence similarity is usually due to evolutionary relationship rather than because it is needed for a particular fold